publications
2025
-
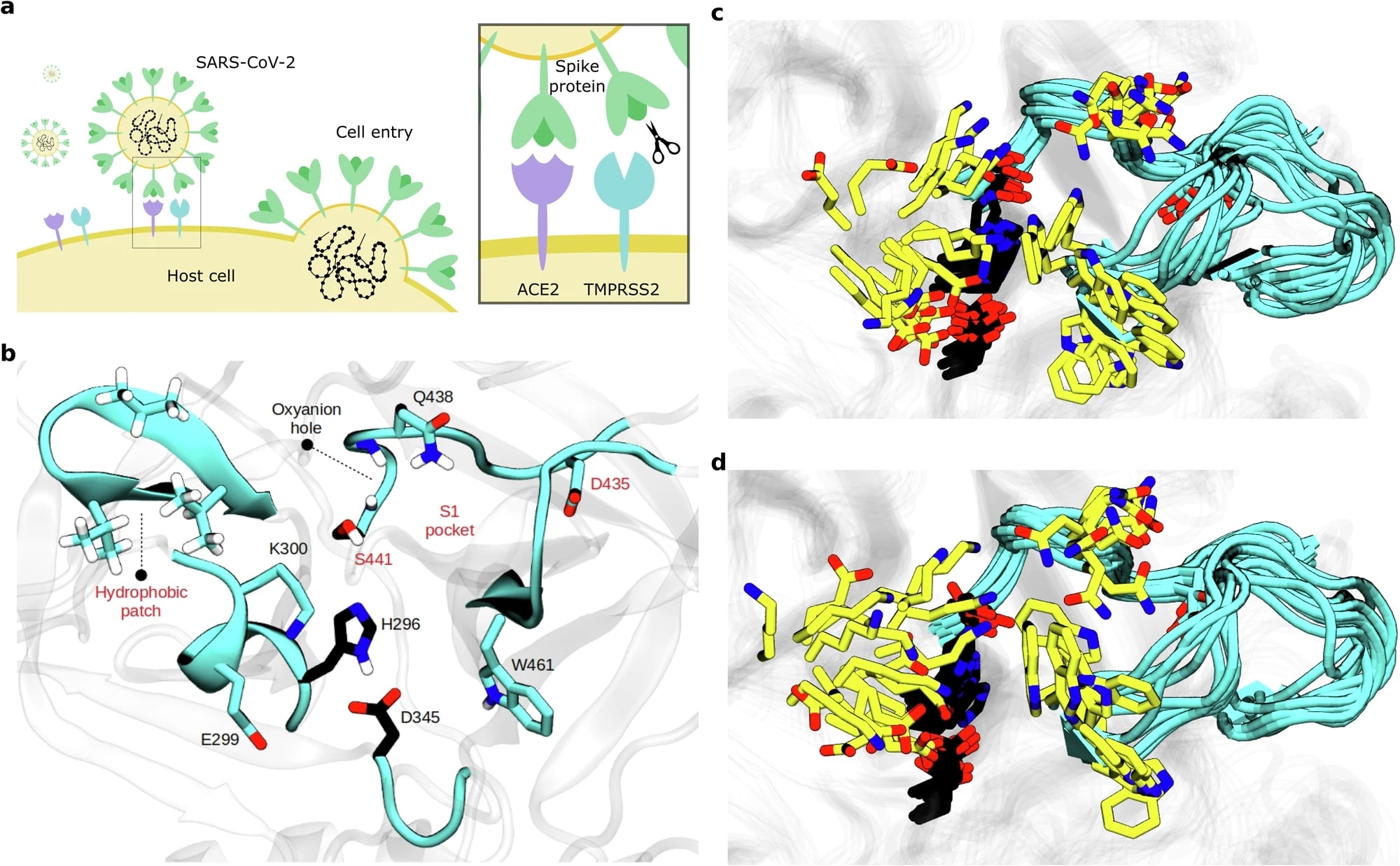 Simulations and active learning enable efficient identification of an experimentally-validated broad coronavirus inhibitorKatarina Elez, Tim Hempel, Jonathan H. Shrimp, Nicole Moor, Lluís Raich, and 7 more authorsNature Communications, 2025
Simulations and active learning enable efficient identification of an experimentally-validated broad coronavirus inhibitorKatarina Elez, Tim Hempel, Jonathan H. Shrimp, Nicole Moor, Lluís Raich, and 7 more authorsNature Communications, 2025Drug screening resembles finding a needle in a haystack: identifying a few effective inhibitors from a large pool of potential drugs. Large experimental screens are expensive and time-consuming, while virtual screening trades off computational efficiency and experimental correlation. Here we develop a framework that combines molecular dynamics (MD) simulations with active learning. Two components drastically reduce the number of candidates needing experimental testing to less than 20: (1) a target-specific score that evaluates target inhibition and (2) extensive MD simulations to generate a receptor ensemble. The active learning approach reduces the number of compounds requiring experimental testing to less than 10 and cuts computational costs by ∼29-fold. Using this framework, we discovered BMS-262084 as a potent inhibitor of TMPRSS2 (IC50 = 1.82 nM). Cell-based experiments confirmed BMS-262084’s efficacy in blocking entry of various SARS-CoV-2 variants and other coronaviruses. The identified inhibitor holds promise for treating viral and other diseases involving TMPRSS2.
@article{elez_simulations_2025, title = {Simulations and active learning enable efficient identification of an experimentally-validated broad coronavirus inhibitor}, author = {Elez, Katarina and Hempel, Tim and Shrimp, Jonathan H. and Moor, Nicole and Raich, Lluís and Rocha, Cheila and Winter, Robin and Le, Tuan and P\"{o}hlmann, Stefan and Hoffmann, Markus and Hall, Matthew D. and Noé, Frank}, year = {2025}, journal = {Nature Communications}, volume = {16}, pages = {6949}, doi = {10.1038/s41467-025-62139-5}, altmetric = true, dimensions = true, bibtex_show = true, selected = true }

2024
-
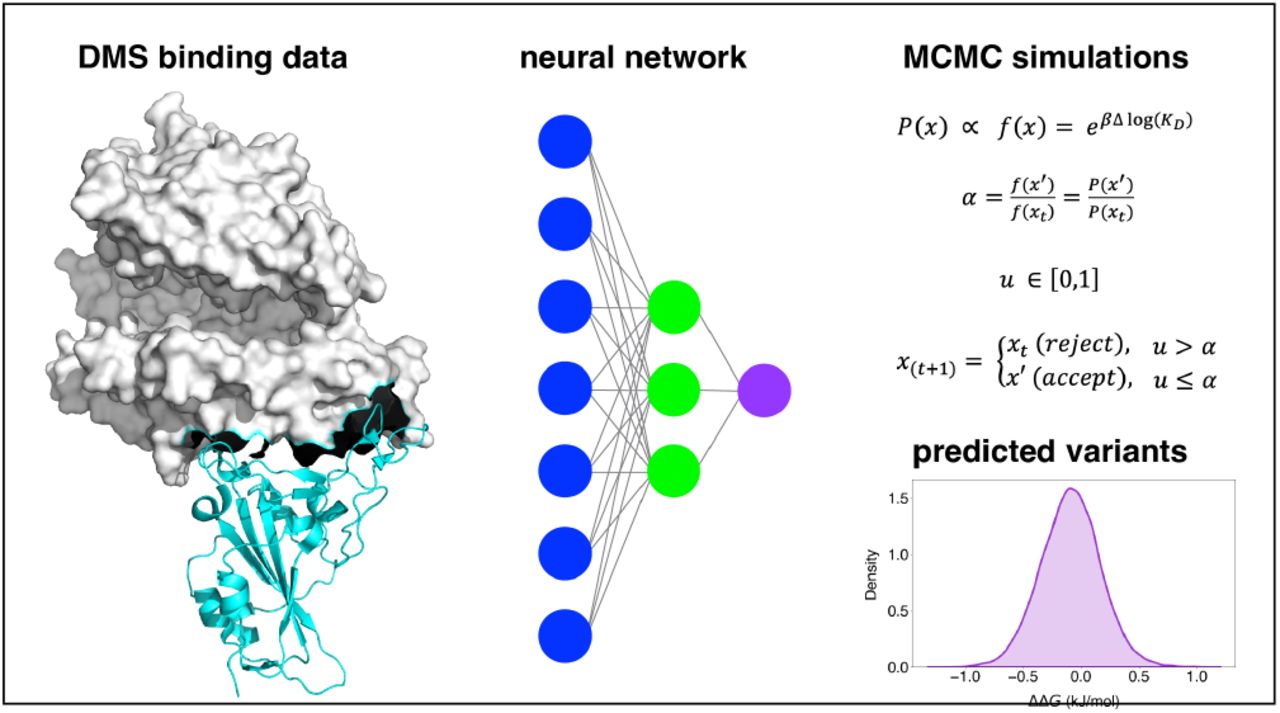 Machine Learning Driven Simulations of SARS-CoV-2 Fitness LandscapeAleksander E. P. Durumeric, Jonas Köhler, Katarina Elez, Lluís Raich, Patricia A. Suriana, and 1 more authorbioRxiv, 2024
Machine Learning Driven Simulations of SARS-CoV-2 Fitness LandscapeAleksander E. P. Durumeric, Jonas Köhler, Katarina Elez, Lluís Raich, Patricia A. Suriana, and 1 more authorbioRxiv, 2024SARS-CoV-2 infection is mediated by interactions between the receptor binding domain (RBD) of viral spike proteins and host cell angiotensin converting enzyme 2 (ACE2) receptors. Mutations in the spike protein are the primary cause for neutralizing antibody escape leading to breakthrough infections. We characterize the fitness landscape underpinning future variants of concern by combining supervised machinelearning and Markov Chain Monte Carlo. Leveraging deep mutational scanning (DMS) data characterizing the binding affinity between RBD mutants to the ACE2 receptor, we predict variants of concern not seen in the training data and sample statistics of the fitness landscape. These simulations provide insight into the relationship between RBD sequence elements and offer a new perspective on utilizing DMS to predict emerging viral strains.
@article{durumeric_machine_2024, title = {Machine {{Learning Driven Simulations}} of {{SARS-CoV-2 Fitness Landscape}}}, author = {Durumeric, Aleksander E. P. and K{\"o}hler, Jonas and Elez, Katarina and Raich, Llu{\'i}s and Suriana, Patricia A. and Sztain, Terra}, year = {2024}, journal = {bioRxiv}, pages = {2024.09.20.614179}, doi = {10.1101/2024.09.20.614179}, altmetric = true, dimensions = true, bibtex_show = true, }

2023
-
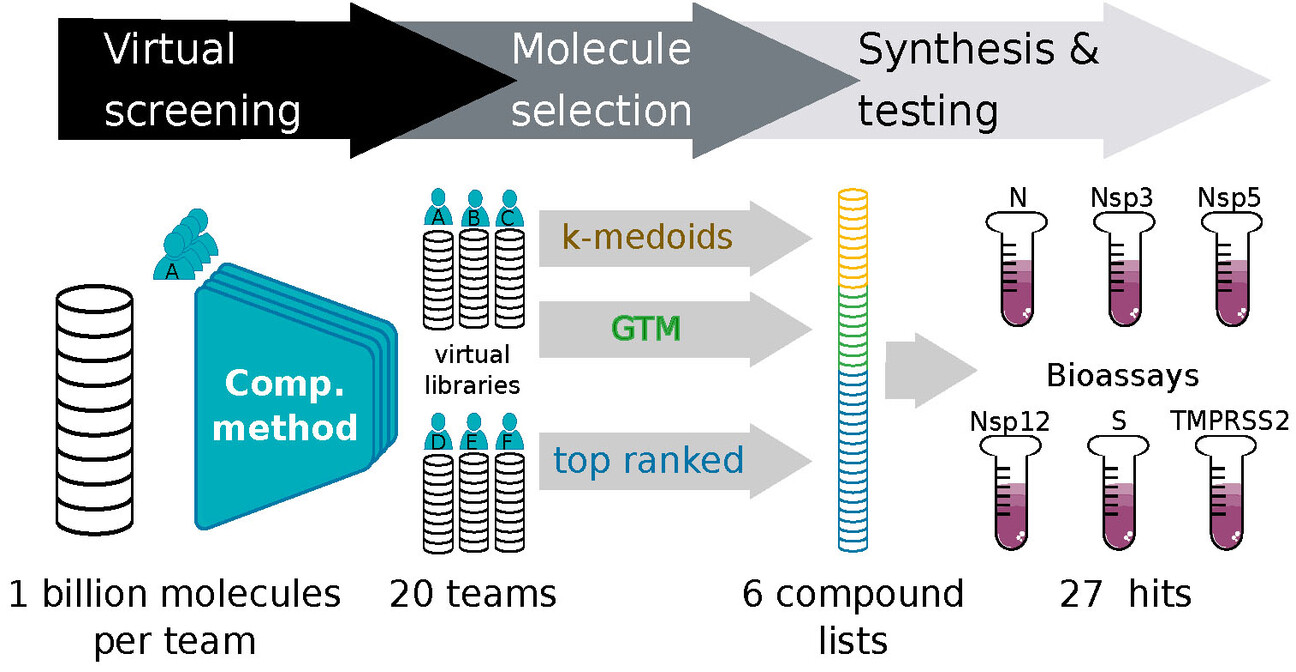 A Community Effort in SARS-CoV-2 Drug DiscoveryJohannes Schimunek, Philipp Seidl, Katarina Elez, Tim Hempel, Tuan Le, and 147 more authorsMolecular Informatics, 2023
A Community Effort in SARS-CoV-2 Drug DiscoveryJohannes Schimunek, Philipp Seidl, Katarina Elez, Tim Hempel, Tuan Le, and 147 more authorsMolecular Informatics, 2023The COVID-19 pandemic continues to pose a substantial threat to human lives and is likely to do so for years to come. Despite the availability of vaccines, searching for efficient small-molecule drugs that are widely available, including in low- and middle-income countries, is an ongoing challenge. In this work, we report the results of an open science community effort, the “Billion molecules against COVID-19 challenge”, to identify small-molecule inhibitors against SARS-CoV-2 or relevant human receptors. Participating teams used a wide variety of computational methods to screen a minimum of 1 billion virtual molecules against 6 protein targets. Overall, 31 teams participated, and they suggested a total of 639,024 molecules, which were subsequently ranked to find ‘consensus compounds’. The organizing team coordinated with various contract research organizations (CROs) and collaborating institutions to synthesize and test 878 compounds for biological activity against proteases (Nsp5, Nsp3, TMPRSS2), nucleocapsid N, RdRP (only the Nsp12 domain), and (alpha) spike protein S. Overall, 27 compounds with weak inhibition/binding were experimentally identified by binding-, cleavage-, and/or viral suppression assays and are presented here. Open science approaches such as the one presented here contribute to the knowledge base of future drug discovery efforts in finding better SARS-CoV-2 treatments.
@article{schimunek_community_2023, title = {A Community Effort in {{SARS-CoV-2}} Drug Discovery}, author = {Schimunek, Johannes and Seidl, Philipp and Elez, Katarina and Hempel, Tim and Le, Tuan and No{\'e}, Frank and Olsson, Simon and Raich, Llu{\'i}s and Winter, Robin and Gokcan, Hatice and Gusev, Filipp and Gutkin, Evgeny M. and Isayev, Olexandr and Kurnikova, Maria G. and Narangoda, Chamali H. and Zubatyuk, Roman and Bosko, Ivan P. and Furs, Konstantin V. and Karpenko, Anna D. and Kornoushenko, Yury V. and Shuldau, Mikita and Yushkevich, Artsemi and Benabderrahmane, Mohammed B. and {Bousquet-Melou}, Patrick and Bureau, Ronan and Charton, Beatrice and Cirou, Bertrand C. and Gil, G{\'e}rard and Allen, William J. and Sirimulla, Suman and Watowich, Stanley and Antonopoulos, Nick and Epitropakis, Nikolaos and Krasoulis, Agamemnon and Pitsikalis, Vassilis and Theodorakis, Stavros and Kozlovskii, Igor and Maliutin, Anton and Medvedev, Alexander and Popov, Petr and Zaretckii, Mark and {Eghbal-Zadeh}, Hamid and Halmich, Christina and Hochreiter, Sepp and Mayr, Andreas and Ruch, Peter and Widrich, Michael and Berenger, Francois and Kumar, Ashutosh and Yamanishi, Yoshihiro and Zhang, Kam Y. J. and Bengio, Emmanuel and Bengio, Yoshua and Jain, Moksh J. and Korablyov, Maksym and Liu, Cheng-Hao and Marcou, Gilles and Glaab, Enrico and Barnsley, Kelly and Iyengar, Suhasini M. and Ondrechen, Mary Jo and Haupt, V. Joachim and Kaiser, Florian and Schroeder, Michael and Pugliese, Luisa and Albani, Simone and Athanasiou, Christina and Beccari, Andrea and Carloni, Paolo and D'Arrigo, Giulia and Gianquinto, Eleonora and Go{\ss}en, Jonas and Hanke, Anton and Joseph, Benjamin P. and Kokh, Daria B. and Kovachka, Sandra and Manelfi, Candida and Mukherjee, Goutam and {Mu{\~n}iz-Chicharro}, Abraham and Musiani, Francesco and {Nunes-Alves}, Ariane and Paiardi, Giulia and Rossetti, Giulia and Sadiq, S. Kashif and Spyrakis, Francesca and Talarico, Carmine and Tsengenes, Alexandros and Wade, Rebecca C. and Copeland, Conner and Gaiser, Jeremiah and Olson, Daniel R. and Roy, Amitava and Venkatraman, Vishwesh and Wheeler, Travis J. and Arthanari, Haribabu and Blaschitz, Klara and Cespugli, Marco and Durmaz, Vedat and Fackeldey, Konstantin and Fischer, Patrick D. and Gorgulla, Christoph and Gruber, Christian and Gruber, Karl and Hetmann, Michael and Kinney, Jamie E. and Padmanabha Das, Krishna M. and Pandita, Shreya and Singh, Amit and Steinkellner, Georg and Tesseyre, Guilhem and Wagner, Gerhard and Wang, Zi-Fu and Yust, Ryan J. and Druzhilovskiy, Dmitry S. and Filimonov, Dmitry A. and Pogodin, Pavel V. and Poroikov, Vladimir and Rudik, Anastassia V. and Stolbov, Leonid A. and Veselovsky, Alexander V. and De Rosa, Maria and De Simone, Giada and Gulotta, Maria R. and Lombino, Jessica and Mekni, Nedra and Perricone, Ugo and Casini, Arturo and Embree, Amanda and Gordon, D. Benjamin and Lei, David and Pratt, Katelin and Voigt, Christopher A. and Chen, Kuang-Yu and Jacob, Yves and Krischuns, Tim and Lafaye, Pierre and Zettor, Agn{\`e}s and Rodr{\'i}guez, M. Luis and White, Kris M. and Fearon, Daren and Von Delft, Frank and Walsh, Martin A. and Horvath, Dragos and Brooks III, Charles L. and Falsafi, Babak and Ford, Bryan and {Garc{\'i}a-Sastre}, Adolfo and Yup Lee, Sang and Naffakh, Nadia and Varnek, Alexandre and Klambauer, G{\"u}nter and Hermans, Thomas M.}, year = {2023}, journal = {Molecular Informatics}, volume = {43}, number = {1}, pages = {e202300262}, doi = {10.1002/minf.202300262}, altmetric = true, dimensions = true, bibtex_show = true, } -
 Pharmaceutical Composition for Treating Covid-19 Comprising Otamixaban and at Least One of Camostat and NafamostatTim Hempel, Katarina Elez, Lluís Raich, Frank Noé, Nadine Krüger, and 2 more authors2023EP4122461A1
Pharmaceutical Composition for Treating Covid-19 Comprising Otamixaban and at Least One of Camostat and NafamostatTim Hempel, Katarina Elez, Lluís Raich, Frank Noé, Nadine Krüger, and 2 more authors2023EP4122461A1The invention relates to a pharmaceutical composition that is appropriate to treat or prevent SARS-CoV-2-related diseases such as COVID-19. The pharmaceutical composition comprises a pharmaceutically effective amount of i) otamixaban or a pharmaceutically acceptable salt thereof and ii) at least one of camostat, a pharmaceutically acceptable salt of camostat, nafamostat, and a pharmaceutically acceptable salt of nafamostat.
@patent{hempel_pharmaceutical_2023, title = {Pharmaceutical {{Composition}} for {{Treating Covid-19 Comprising Otamixaban}} and at {{Least One}} of {{Camostat}} and {{Nafamostat}}}, author = {Hempel, Tim and Elez, Katarina and Raich, Llu{\'i}s and No{\'e}, Frank and Kr{\"u}ger, Nadine and Hoffmann, Markus and P{\"o}hlmann, Stefan}, year = {2023}, number = {EP4122461A1}, assignee = {Freie Universitaet Berlin, Deutsches Primatenzentrum GmbH}, note = {EP4122461A1}, bibtex_show = true, }


2021
-
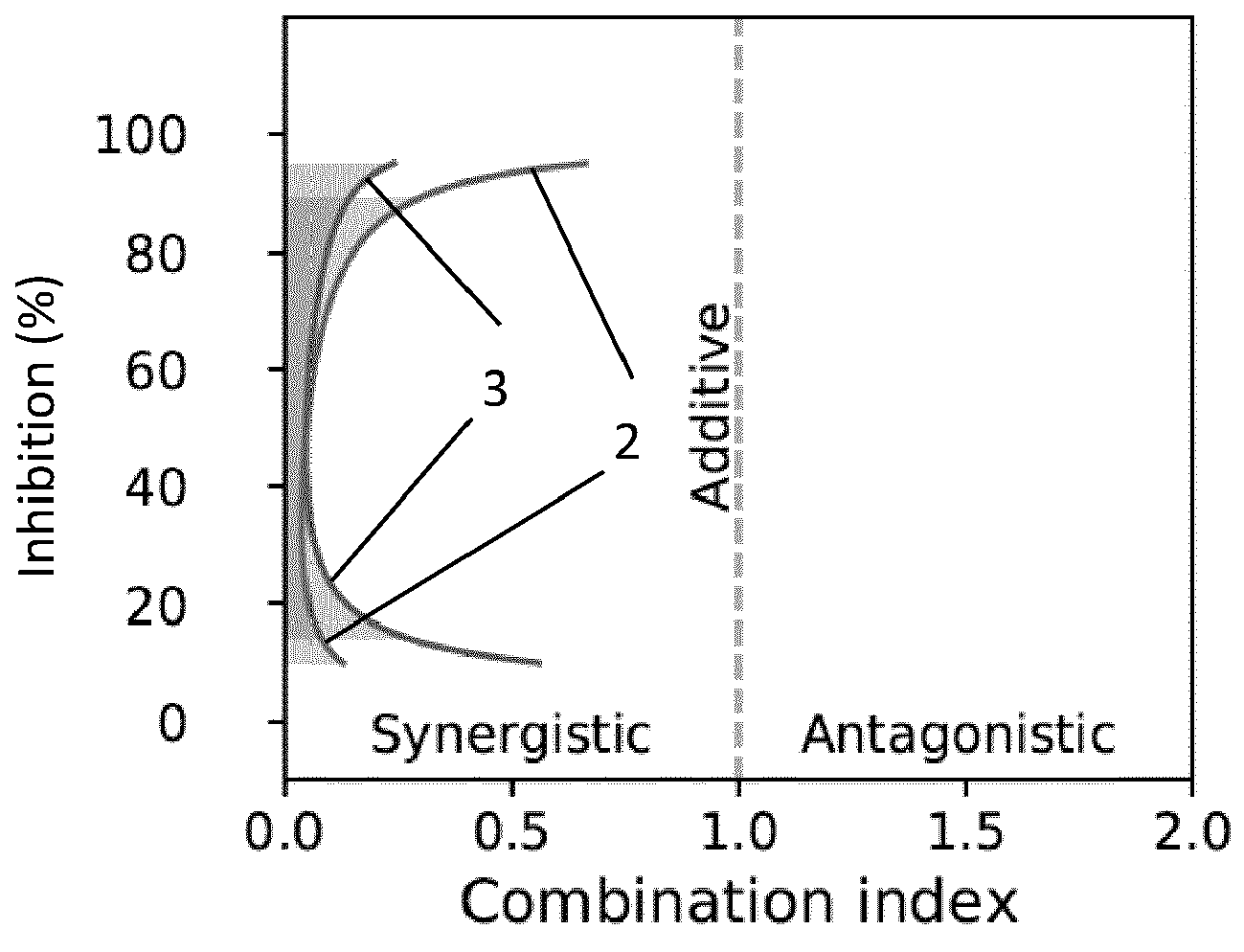
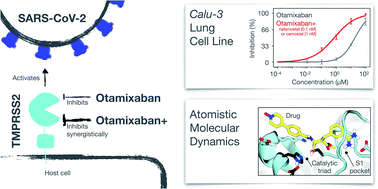 Synergistic Inhibition of SARS-CoV-2 Cell Entry by Otamixaban and Covalent Protease Inhibitors: Pre-Clinical Assessment of Pharmacological and Molecular PropertiesTim Hempel, Katarina Elez, Nadine Krüger, Lluís Raich, Jonathan H. Shrimp, and 8 more authorsChemical Science, 2021
Synergistic Inhibition of SARS-CoV-2 Cell Entry by Otamixaban and Covalent Protease Inhibitors: Pre-Clinical Assessment of Pharmacological and Molecular PropertiesTim Hempel, Katarina Elez, Nadine Krüger, Lluís Raich, Jonathan H. Shrimp, and 8 more authorsChemical Science, 2021SARS-CoV-2, the cause of the COVID-19 pandemic, exploits host cell proteins for viral entry into human lung cells. One of them, the protease TMPRSS2, is required to activate the viral spike protein (S). Even though two inhibitors, camostat and nafamostat, are known to inhibit TMPRSS2 and block cell entry of SARS-CoV-2, finding further potent therapeutic options is still an important task. In this study, we report that a late-stage drug candidate, otamixaban, inhibits SARS-CoV-2 cell entry. We show that otamixaban suppresses TMPRSS2 activity and SARS-CoV-2 infection of a human lung cell line, although with lower potency than camostat or nafamostat. In contrast, otamixaban inhibits SARS-CoV-2 infection of precision cut lung slices with the same potency as camostat. Furthermore, we report that otamixaban’s potency can be significantly enhanced by (sub-) nanomolar nafamostat or camostat supplementation. Dominant molecular TMPRSS2-otamixaban interactions are assessed by extensive 109 μs of atomistic molecular dynamics simulations. Our findings suggest that combinations of otamixaban with supplemental camostat or nafamostat are a promising option for the treatment of COVID-19.
@article{hempel_synergistic_2021, title = {Synergistic Inhibition of {{SARS-CoV-2}} Cell Entry by Otamixaban and Covalent Protease Inhibitors: Pre-Clinical Assessment of Pharmacological and Molecular Properties}, author = {Hempel, Tim and Elez, Katarina and Kr{\"u}ger, Nadine and Raich, Llu{\'i}s and H.~Shrimp, Jonathan and Danov, Olga and Jonigk, Danny and Braun, Armin and Shen, Min and D.~Hall, Matthew and P{\"o}hlmann, Stefan and Hoffmann, Markus and No{\'e}, Frank}, year = {2021}, journal = {Chemical Science}, volume = {12}, number = {38}, pages = {12600--12609}, publisher = {Royal Society of Chemistry}, doi = {10.1039/D1SC01494C}, altmetric = true, dimensions = true, bibtex_show = true, selected = true } -
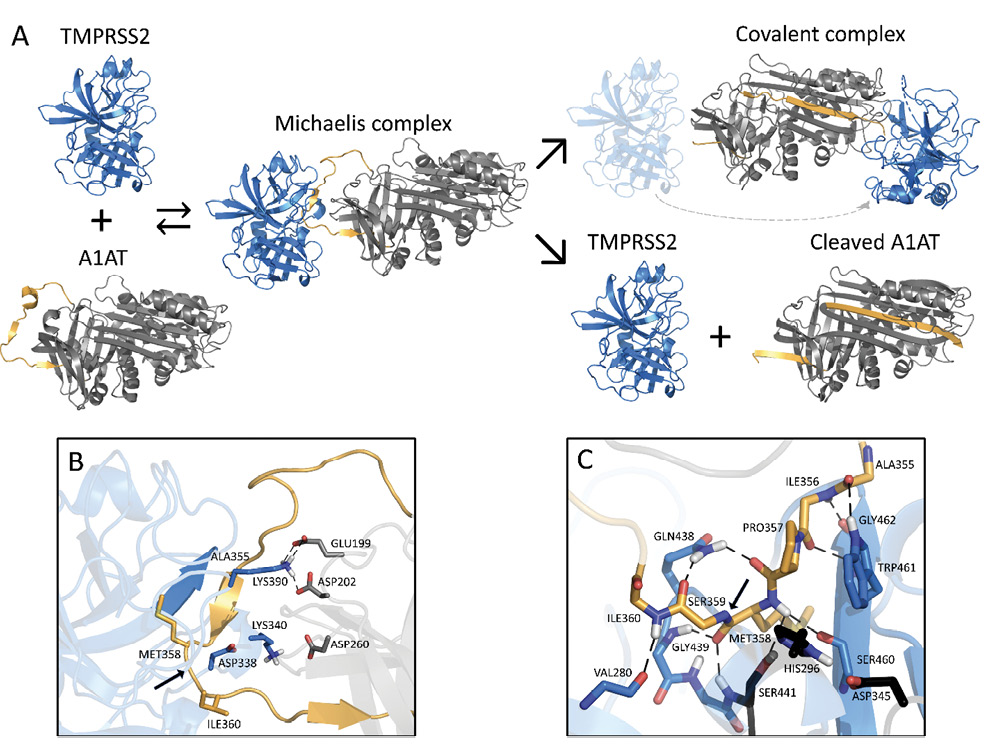 Alpha 1 Antitrypsin Is an Inhibitor of the SARS-CoV-2–Priming Protease TMPRSS2Nurit P. Azouz, Andrea Klingler, Victoria Callahan, Ivan Akhrymuk, Katarina Elez, and 7 more authorsPathogens and Immunity, 2021
Alpha 1 Antitrypsin Is an Inhibitor of the SARS-CoV-2–Priming Protease TMPRSS2Nurit P. Azouz, Andrea Klingler, Victoria Callahan, Ivan Akhrymuk, Katarina Elez, and 7 more authorsPathogens and Immunity, 2021Background: Host proteases have been suggested to be crucial for dissemination of MERS, SARS-CoV, and SARS-CoV-2 coronaviruses, but the relative contribution of membrane versus intracellular proteases remains controversial. Transmembrane serine protease 2 (TMPRSS2) is regarded as one of the main proteases implicated in the coronavirus S protein priming, an important step for binding of the S protein to the angiotensin-converting enzyme 2 (ACE2) receptor before cell entry. Methods: We developed a cell-based assay to identify TMPRSS2 inhibitors. Inhibitory activity was established in SARS-CoV-2 viral load systems. Results: We identified the human extracellular serine protease inhibitor (serpin) alpha 1 antitrypsin (A1AT) as a novel TMPRSS2 inhibitor. Structural modeling revealed that A1AT docked to an extracellular domain of TMPRSS2 in a conformation that is suitable for catalysis, resembling similar serine protease inhibitor complexes. Inhibitory activity of A1AT was established in a SARS-CoV-2 viral load system. Notably, plasma A1AT levels were associated with COVID-19 disease severity. Conclusions: Our data support the key role of extracellular serine proteases in SARS CoV-2 infections and indicate that treatment with serpins, particularly the FDA-approved drug A1AT, may be effective in limiting SARS-CoV-2 dissemination by affecting the surface of the host cells.
@article{azouz_alpha_2021, title = {Alpha 1 {{Antitrypsin}} Is an {{Inhibitor}} of the {{SARS-CoV-2}}--{{Priming Protease TMPRSS2}}}, author = {Azouz, Nurit P. and Klingler, Andrea and Callahan, Victoria and Akhrymuk, Ivan and Elez, Katarina and Raich, Llu{\'i}s and Henry, Brandon and Benoit, Justin and Benoit, Stefanie and No{\'e}, Frank and {Kehn-Hall}, Kylene and Rothenberg, Marc}, year = {2021}, journal = {Pathogens and Immunity}, volume = {6}, number = {1}, pages = {55--74}, doi = {10.20411/pai.v6i1.408}, altmetric = true, dimensions = true, bibtex_show = true, }


2020
-
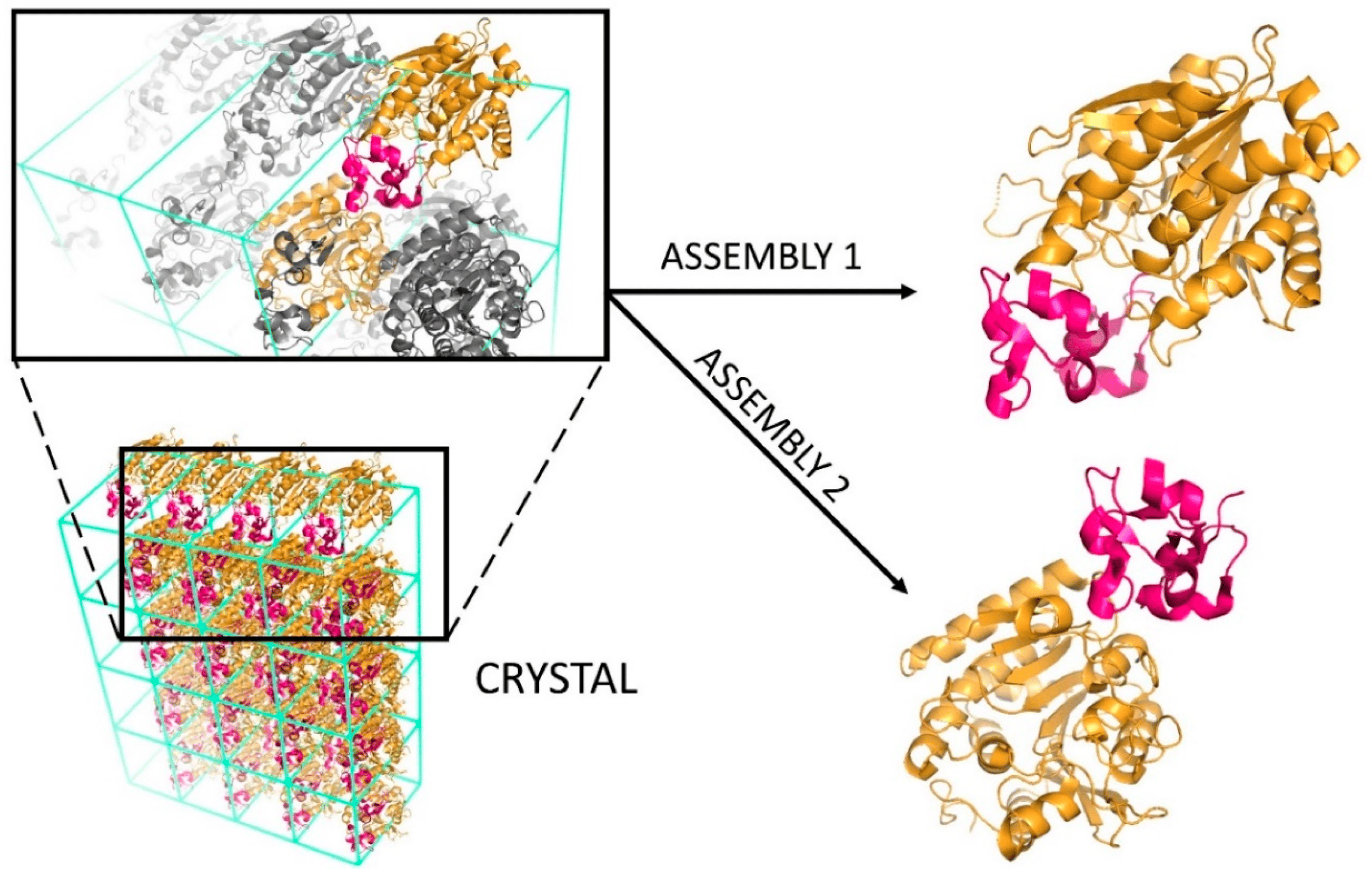 Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their ClassificationKatarina Elez, Alexandre M. J. J. Bonvin, and Anna VangoneCrystals, 2020
Biological vs. Crystallographic Protein Interfaces: An Overview of Computational Approaches for Their ClassificationKatarina Elez, Alexandre M. J. J. Bonvin, and Anna VangoneCrystals, 2020Complexes between proteins are at the basis of almost every process in cells. Their study, from a structural perspective, has a pivotal role in understanding biological functions and, importantly, in drug development. X-ray crystallography represents the broadest source for the experimental structural characterization of protein-protein complexes. Correctly identifying the biologically relevant interface from the crystallographic ones is, however, not trivial and can be prone to errors. Over the past two decades, computational methodologies have been developed to study the differences of those interfaces and automatically classify them as biological or crystallographic. Overall, protein-protein interfaces show differences in terms of composition, energetics and evolutionary conservation between biological and crystallographic ones. Based on those observations, a number of computational methods have been developed for this classification problem, which can be grouped into three main categories: Energy-, empirical knowledge- and machine learning-based approaches. In this review, we give a comprehensive overview of the training datasets and methods so far implemented, providing useful links and a brief description of each method.
@article{elez_biological_2020, title = {Biological vs. {{Crystallographic Protein Interfaces}}: {{An Overview}} of {{Computational Approaches}} for {{Their Classification}}}, shorttitle = {Biological vs. {{Crystallographic Protein Interfaces}}}, author = {Elez, Katarina and Bonvin, Alexandre M. J. J. and Vangone, Anna}, year = {2020}, journal = {Crystals}, volume = {10}, number = {2}, pages = {114}, publisher = {Multidisciplinary Digital Publishing Institute}, doi = {10.3390/cryst10020114}, altmetric = true, dimensions = true, bibtex_show = true, }

2019
-
 PRODIGY-crystal: A Web-Tool for Classification of Biological Interfaces in Protein ComplexesBrian Jiménez-García, Katarina Elez, Panagiotis I Koukos, Alexandre Mjj Bonvin, and Anna VangoneBioinformatics, 2019
PRODIGY-crystal: A Web-Tool for Classification of Biological Interfaces in Protein ComplexesBrian Jiménez-García, Katarina Elez, Panagiotis I Koukos, Alexandre Mjj Bonvin, and Anna VangoneBioinformatics, 2019Distinguishing biologically relevant interfaces from crystallographic ones in biological complexes is fundamental in order to associate cellular functions to the correct macromolecular assemblies. Recently, we described a detailed study reporting the differences in the type of intermolecular residue–residue contacts between biological and crystallographic interfaces. Our findings allowed us to develop a fast predictor of biological interfaces reaching an accuracy of 0.92 and competitive to the current state of the art. Here we present its web-server implementation, PRODIGY-CRYSTAL, aimed at the classification of biological and crystallographic interfaces. PRODIGY-CRYSTAL has the advantage of being fast, accurate and simple. This, together with its user-friendly interface and user support forum, ensures its broad accessibility.PRODIGY-CRYSTAL is freely available without registration requirements at https://haddock.science.uu.nl/services/PRODIGY-CRYSTAL.
@article{jimenez-garcia_prodigycrystal_2019, title = {{{PRODIGY-crystal}}: A Web-Tool for Classification of Biological Interfaces in Protein Complexes}, shorttitle = {{{PRODIGY-crystal}}}, author = {{Jim{\'e}nez-Garc{\'i}a}, Brian and Elez, Katarina and Koukos, Panagiotis I and Bonvin, Alexandre Mjj and Vangone, Anna}, year = {2019}, journal = {Bioinformatics}, volume = {35}, number = {22}, pages = {4821--4823}, doi = {10.1093/bioinformatics/btz437}, altmetric = true, dimensions = true, bibtex_show = true, }

2018
-
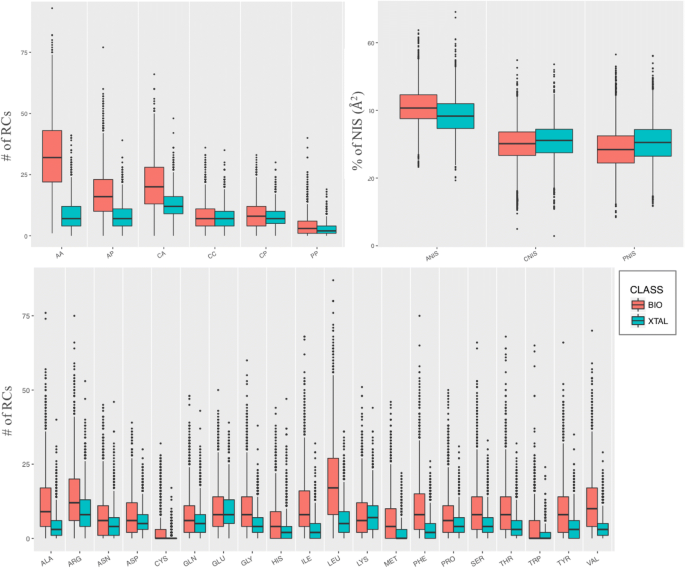 Distinguishing Crystallographic from Biological Interfaces in Protein Complexes: Role of Intermolecular Contacts and Energetics for ClassificationKatarina Elez, Alexandre M. J. J. Bonvin, and Anna VangoneBMC Bioinformatics, 2018
Distinguishing Crystallographic from Biological Interfaces in Protein Complexes: Role of Intermolecular Contacts and Energetics for ClassificationKatarina Elez, Alexandre M. J. J. Bonvin, and Anna VangoneBMC Bioinformatics, 2018Study of macromolecular assemblies is fundamental to understand functions in cells. X-ray crystallography is the most common technique to solve their 3D structure at atomic resolution. In a crystal, however, both biologically-relevant interfaces and non-specific interfaces resulting from crystallographic packing are observed. Due to the complexity of the biological assemblies currently tackled, classifying those interfaces, i.e. distinguishing biological from crystal lattice interfaces, is not trivial and often prone to errors. In this context, analyzing the physico-chemical characteristics of biological/crystal interfaces can help researchers identify possible features that distinguish them and gain a better understanding of the systems.
@article{elez_distinguishing_2018, title = {Distinguishing Crystallographic from Biological Interfaces in Protein Complexes: Role of Intermolecular Contacts and Energetics for Classification}, shorttitle = {Distinguishing Crystallographic from Biological Interfaces in Protein Complexes}, author = {Elez, Katarina and Bonvin, Alexandre M. J. J. and Vangone, Anna}, year = {2018}, journal = {BMC Bioinformatics}, volume = {19}, number = {15}, pages = {438}, doi = {10.1186/s12859-018-2414-9}, altmetric = true, dimensions = true, bibtex_show = true, selected = true }

2017
-
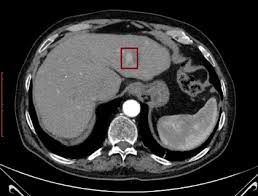 A Novel Approach for Hepatocellular Carcinoma Detection and Classification Based on Triphasic CT ProtocolVitoantonio Bevilacqua, Antonio Brunetti, Gianpaolo Francesco Trotta, Giovanni Dimauro, Katarina Elez, and 2 more authorsIn 2017 IEEE Congress on Evolutionary Computation (CEC), 2017
A Novel Approach for Hepatocellular Carcinoma Detection and Classification Based on Triphasic CT ProtocolVitoantonio Bevilacqua, Antonio Brunetti, Gianpaolo Francesco Trotta, Giovanni Dimauro, Katarina Elez, and 2 more authorsIn 2017 IEEE Congress on Evolutionary Computation (CEC), 2017Introduction and objective: Computer Aided Decision (CAD) systems based on Medical Imaging could support radiologists in grading Hepatocellular carcinoma (HCC) by means of Computed Tomography (CT) images, avoiding medical invasive procedures such as biopsies. The identification and characterization of Regions of Interest (ROIs) containing lesions is an important phase allowing an easier classification in two classes of HCCs. Two steps are needed for the detection of lesioned ROIs: a liver isolation in each CT slice and a lesion segmentation. Materials and methods: In our previous study, materials consisted in abdominal CT hepatic lesions of only three patients subjected to liver transplant, partial hepatectomy, or US-guided needle biopsy. In this paper, thanks to a more extensively phase of data collection, available materials impressively grew to 18 patients belonging to 2-balanced classes. Several approaches were implemented to segment the region of liver and, then, to detect the ROI of the lesions. At the end of these preprocessing phases, we extracted the same morphological features of the previous work and designed an evolutionary algorithm to optimize neural network classifiers based on different subsets of features. Results and conclusion: Tests conducted on the new ANN topologies showed a higher generalization of the average performance indices regardless of the applied training, validation and test sets, confirming both the validity and the robustness of the approach of previous study even though the limited number of patients.
@inproceedings{bevilacqua_novel_2017, title = {A Novel Approach for {{Hepatocellular Carcinoma}} Detection and Classification Based on Triphasic {{CT Protocol}}}, booktitle = {2017 {{IEEE Congress}} on {{Evolutionary Computation}} ({{CEC}})}, author = {Bevilacqua, Vitoantonio and Brunetti, Antonio and Trotta, Gianpaolo Francesco and Dimauro, Giovanni and Elez, Katarina and Alberotanza, Vito and Scardapane, Arnaldo}, year = {2017}, pages = {1856--1863}, doi = {10.1109/CEC.2017.7969527}, altmetric = true, dimensions = true, bibtex_show = true, }
